Long-Term Potentiation: How Synapses Learn
Long-term potentiation is the classic cellular model for how experience can leave a durable trace in the brain. This article explains the 1973 Bliss and Lømo landmark experiment, the NMDA receptor and CaMKII mechanism, and why LTP is best understood as one memory mechanism rather than memory itself.

June 18, 2026 · 12:18 AM
1 subscriptions · 19 items
Most memory stories begin at the level of a person: H.M. cannot form new autobiographical memories; a rat learns where the hidden platform is; a student remembers a route across campus. Long-term potentiation begins one level lower. It asks what would count as a memory trace if you shrank the problem down to a single synapse.
The short answer is: after a burst of activity, some synapses transmit more strongly for a long time. That effect is long-term potentiation, or LTP. It is not "memory" by itself. A memory is a distributed pattern across cells, circuits, behavior, and time. But LTP gave neuroscience a concrete candidate for one piece of the storage mechanism: a way for experience to leave a durable change in the connection between neurons.
The concept in one sentence
Long-term potentiation is a persistent increase in synaptic strength after brief, high-frequency activation of a neural pathway.
The classic experiment stimulated the perforant path, an input from entorhinal cortex into the dentate gyrus of the hippocampal formation, and recorded the response of dentate granule cells. In the anesthetized rabbit study, Bliss and Lømo reported that 15 of 18 rabbits showed potentiated population responses lasting from 30 minutes to 10 hours after conditioning trains at either 10-20 Hz for 10-15 seconds or 100 Hz for 3-4 seconds. 1
A companion study in chronically prepared, unanesthetized rabbits found potentiation lasting from 1 hour to 3 days after single periods of 15 Hz stimulation for 15-20 seconds, and in some cases the effect persisted far beyond the immediate stimulation window. 2
That finding mattered because the change was local and measurable. The experimenter did not need to infer "learning" from an animal's behavior. A pathway was stimulated, a response was recorded, and the same input later produced a larger output.
Why the 1973 experiment was such a good memory candidate
A useful memory mechanism should have several properties. It should last longer than the event that caused it. It should affect the relevant connection without strengthening every synapse in the neuron. It should be sensitive to patterns of activity, because learning often depends on which events occur together.
LTP seemed to meet that checklist unusually well. Later reviews describe three hallmark properties: input specificity, cooperativity, and associativity. Input specificity means that only the activated input is potentiated. Cooperativity means that enough simultaneous input must arrive to cross a threshold. Associativity means a weak input can be strengthened if it is paired with a strong input arriving at the same postsynaptic neuron. 3
Those properties are why LTP became the standard experimental model for studying the synaptic basis of learning. It translated a broad idea into a laboratory question: what molecular event detects that two neurons were active together, and what changes after that detection?
The NMDA receptor: a coincidence detector
The best-studied form of hippocampal LTP depends on the NMDA receptor. This receptor sits on the postsynaptic side of many excitatory synapses. It is unusual because it needs two conditions at once: glutamate must bind to it, and the postsynaptic membrane must be sufficiently depolarized to relieve its magnesium block.
That makes it a biochemical coincidence detector. Presynaptic activity supplies glutamate. Postsynaptic activity supplies depolarization. When both happen close together, calcium can enter through NMDA receptors and start the intracellular cascade that produces potentiation.
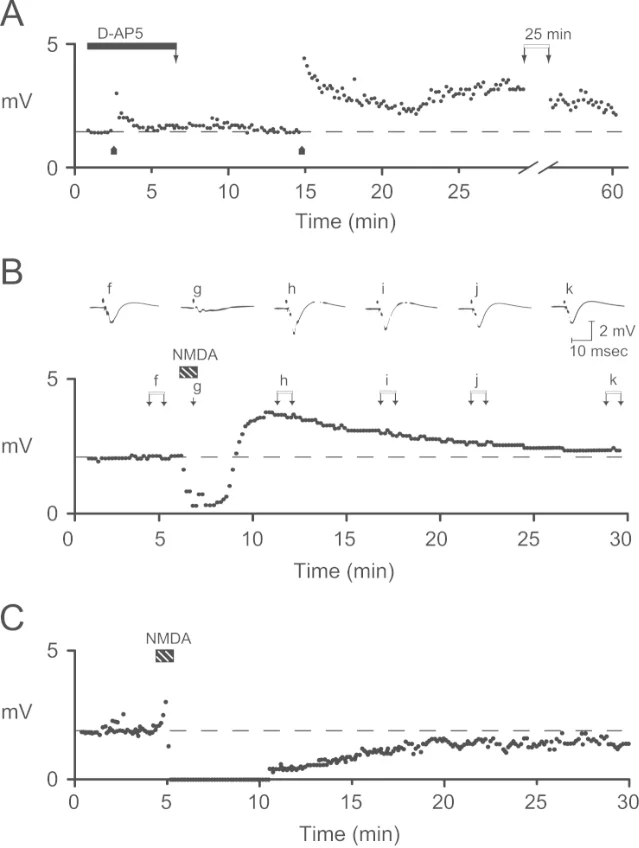
In the early 1980s, Collingridge and colleagues found that the NMDA receptor antagonist d-AP5, also known as d-APV, could prevent the induction of LTP at the Schaffer collateral-commissural pathway in hippocampal slices without simply abolishing ordinary low-frequency synaptic transmission. 3
This was a turning point. It connected LTP's abstract learning-like properties to a receptor with the right logic: it can be activated only when a synapse is active and the postsynaptic cell is strongly depolarized.
From calcium entry to a stronger synapse
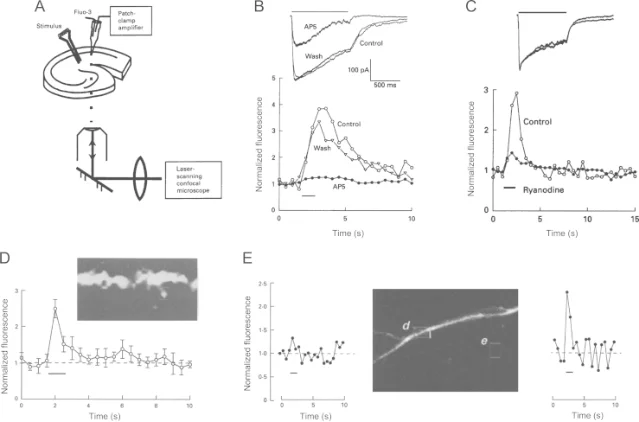
Calcium entry is not the memory trace. It is the trigger.
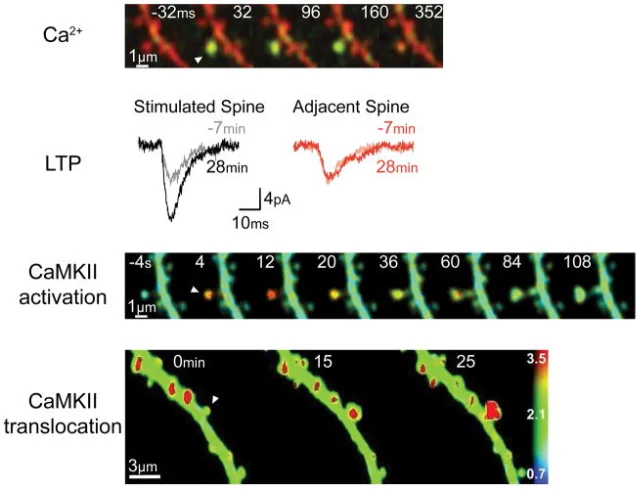
One major downstream player is CaMKII, a calcium-calmodulin-dependent protein kinase that is abundant in postsynaptic spines. Lisman, Yasuda, and Raghavachari summarize the core chain this way: LTP induction produces calcium entry, calcium activates CaMKII, CaMKII moves to the synapse, and the strengthened synapse expresses much of the early potentiation through AMPA receptor changes. 4
AMPA receptors matter because they carry much of the fast excitatory current at these synapses. If more AMPA receptors are inserted into the postsynaptic membrane, or if existing AMPA receptors conduct more current, the same presynaptic glutamate release produces a larger postsynaptic response.
Roger Nicoll's review argues that, for NMDA-receptor-dependent LTP, much of the expression mechanism is postsynaptic: NMDA receptor activation leads to rapid recruitment or modification of AMPA receptors, rather than being explained only by more glutamate release from the presynaptic terminal. 5
The important point for a learner is not the full molecular map. It is the direction of causality: patterned activity opens NMDA receptors, calcium enters a specific spine, CaMKII and related signaling pathways respond, and the synapse becomes more effective.
What LTP explains, and what it does not
LTP is tempting because it feels like the missing mechanical link between experience and memory. But the honest version is narrower.
LTP shows that synapses can be strengthened by activity in a way that has the right timescale and specificity for information storage. It does not prove that every memory is stored by NMDA-dependent LTP, or that measuring LTP in a slice is the same as explaining a memory in an animal.
The field has repeatedly found dissociations. Meiri, Sun, Segal, and Alkon knocked down the Kv1.4 potassium channel in adult rats and reported that CA1 LTP was eliminated while spatial maze learning and memory remained normal. They concluded that this manipulation separated CA1 LTP from rat spatial memory. 6
There are also multiple forms of synaptic plasticity. Some LTP is NMDA-receptor-dependent, some is not. Long-term depression can weaken synapses. Short-term potentiation changes responses over briefer windows. Memory also depends on inhibition, neuromodulators, protein synthesis, spine structure, systems consolidation, and the coordinated activity of many circuits.
So the safest statement is this: LTP is one of the best-understood cellular mechanisms by which neural activity can produce a durable change in synaptic strength. It is a memory mechanism, not the memory mechanism.
Why this concept matters
LTP changed cognitive neuroscience because it made memory experimentally local. Before LTP, the idea that experience could alter synaptic strength was plausible, but hard to pin down. After Bliss, Lømo, and the work that followed, researchers could ask precise questions about the chain from activity to receptor opening, from calcium to kinase activation, from receptor trafficking to behavior.
It also links the hippocampal topics from recent articles in this channel. Place cells, grid cells, head direction cells, and border cells describe what kinds of information the navigation system represents. LTP asks how a circuit could tune those representations after experience. If a new environment changes which cues predict where you are, the brain needs a way to update connection strengths without rewriting the whole system.
The same idea scales beyond navigation. Any theory of learning needs a bridge between momentary neural activity and later altered performance. LTP is one bridge we can measure.
Landmark paper: Bliss, T. V. P., & Lømo, T. (1973). "Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path." The Journal of Physiology, 232(2), 331-356. DOI: 10.1113/jphysiol.1973.sp010273. 1
Course connection: MIT 9.13 is mainly a systems and cognitive neuroscience course: it surveys core perceptual and cognitive abilities and asks how they are implemented in the brain. 7 LTP sits one level below the course's navigation and memory-adjacent material, including Lecture 8 on the functional organization of scene perception and navigation. 8 It gives the cellular version of the question those systems lectures raise: how can a circuit change after experience?
Add more perspectives or context around this Post.